
Dr Matthew Hoare | Understanding Gene Mutations in Chronic Liver Disease
About this episode
The liver plays a vital role in keeping us healthy, by controlling levels of sugars and fats in our blood, as well as clearing the blood of toxins. Chronic liver disease affects around 25% of the population and is reported to be the third largest cause of premature death in the UK. Liver disease can occur as the result of long-term consumption of alcohol or viral hepatitis, but the fastest growing cause is non-alcoholic fatty liver disease, associated with obesity and type 2 diabetes. Read More
Original Article Reference
Summary of the papers ‘Somatic mutations and clonal dynamics in healthy and cirrhotic human liver’, in Nature, https://doi.org/10.1038/s41586-019-1670-9; and ‘Convergent somatic mutations in metabolism genes in chronic liver disease’ in Nature, https://doi.org/10.1038/s41586-021-03974-6
Contact
For further information, you can connect with Dr Matthew Hoare at Matthew.Hoare@cruk.cam.ac.uk
This work is licensed under a Creative Commons Attribution 4.0 International License. 
What does this mean?
Share: You can copy and redistribute the material in any medium or format
Adapt: You can change, and build upon the material for any purpose, even commercially.
Credit: You must give appropriate credit, provide a link to the license, and indicate if changes were made.
Are you ready to increase the impact of your research?
More episodes
What is SciTube? Science Animation for Researchers

IAPB – Seeds of Innovation: The Science Driving Sustainable Agriculture
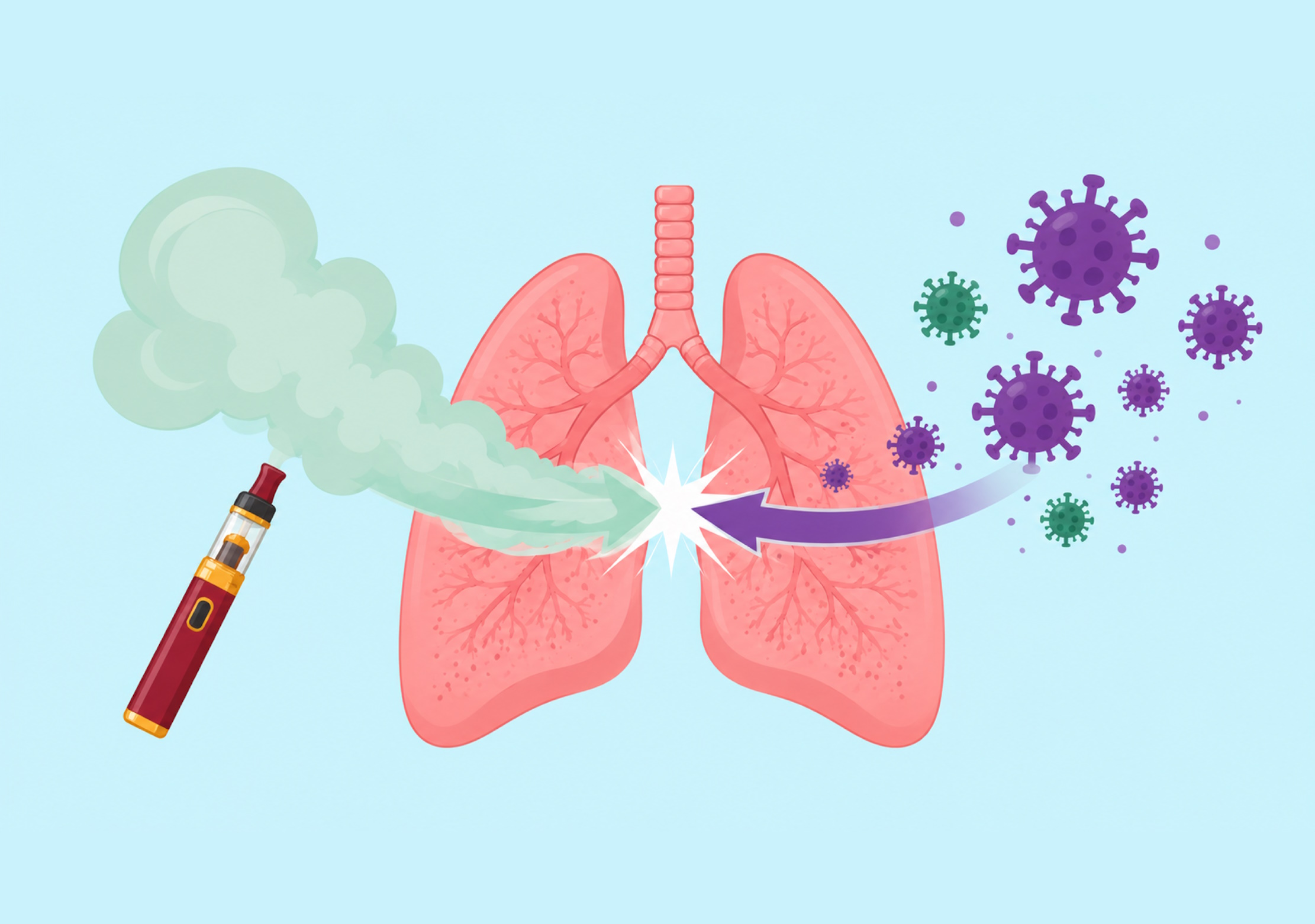
Rob Onyenwoke | How Vaping Can Make Your Lungs More Vulnerable to Infection
Is SciTube Predatory? No. SciTube is a Paid Science Communication Platform
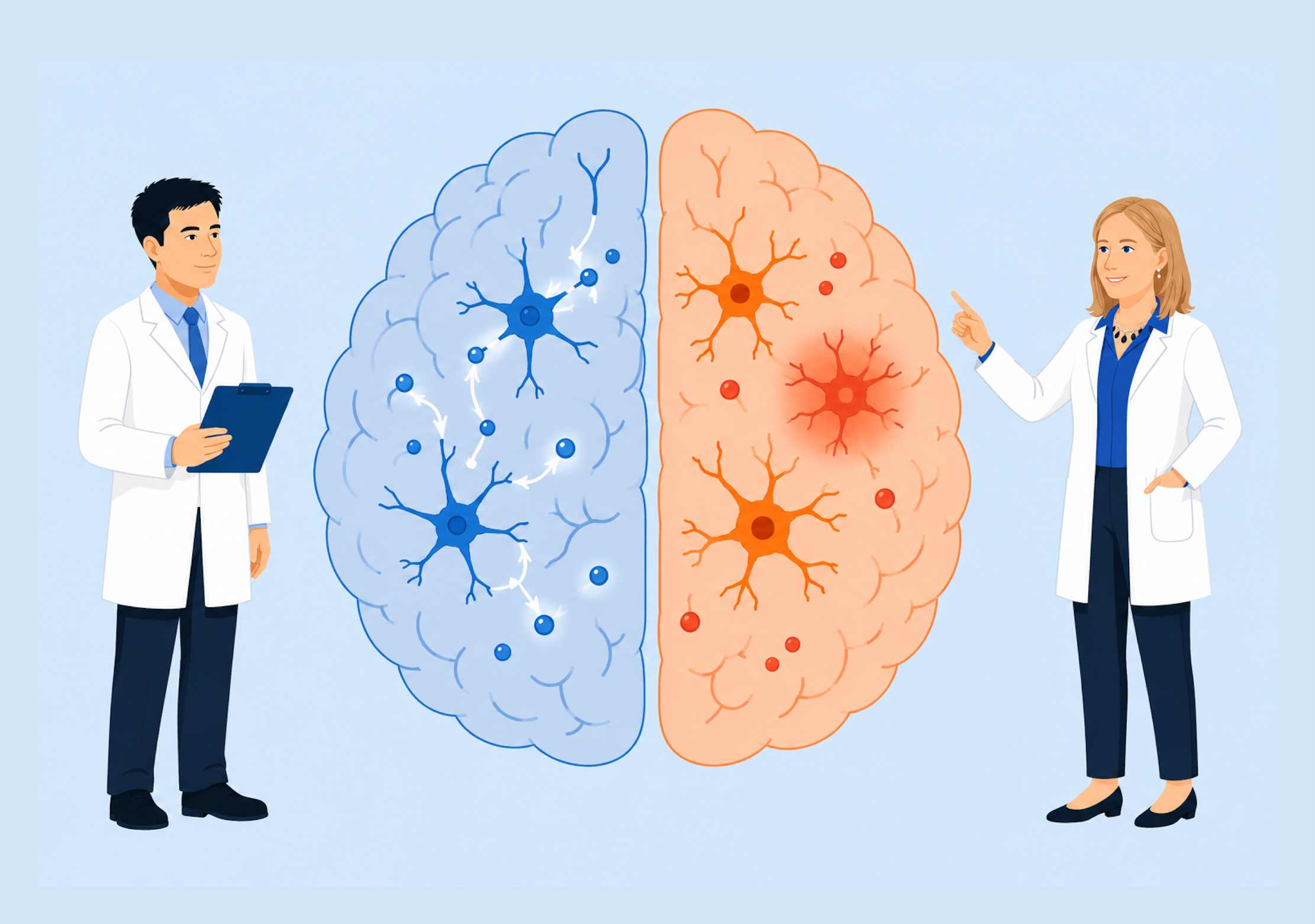
Dr. Xiaolei Zhu – Dr. Barbara Slusher | A New Approach to Depression Treatment Targets Brain Immune Cells
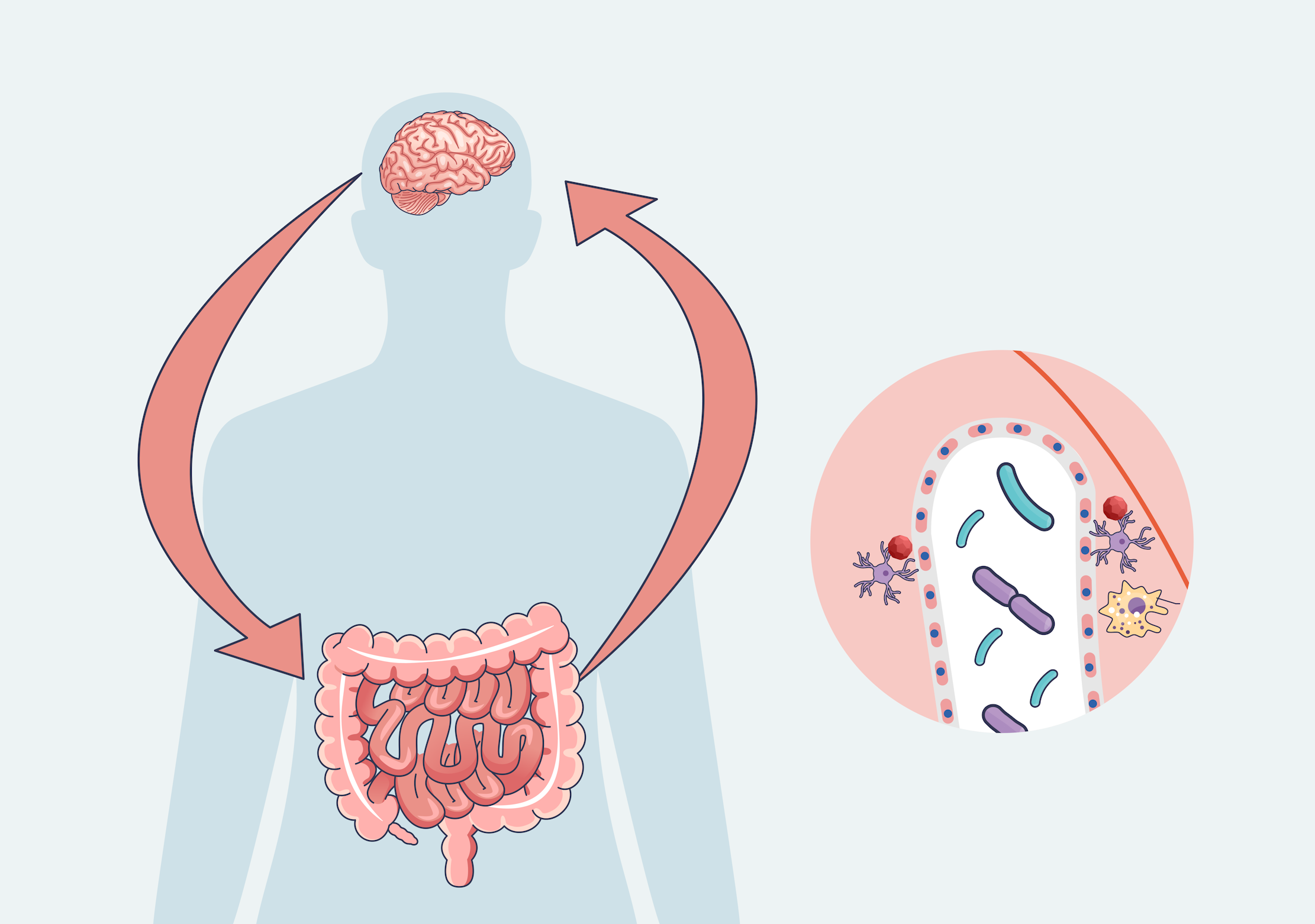
Dr Vedrana Bali – Dr Vladimir Grubišić | How Enteric Glia Shape Health and Disease
Stay Up To Date With SciTube
Subscribe to receive our latest videos straight to your mailbox